1H-1H coupling constants are one of the primary sources of information for nuclear magnetic resonance (NMR) structural analysis. Several selective 2DJ experiments have been proposed that allow for their individual measurement at pure shift resolution. However, all of these experiments fail in the not uncommon case when coupled protons have very close chemical shifts. First, the coupling between protons with overlapping multiplets is inaccessible due to the inability of a frequency-selective pulse to invert just one of them. Second, the strong coupling condition affects the accuracy of coupling measurements involving third spins. These shortcomings impose a limit on the effectiveness of state-of-the-art experiments, such as G-SERF or PSYCHEDELIC. Here, we introduce two new and complementary selective 2DJ experiments that we coin SERFBIRD and SATASERF. These experiments overcome the aforementioned issues by utilizing the 13C satellite signals at natural isotope abundance, which resolves the chemical shift degeneracy. We demonstrate the utility of these experiments on the tetrasaccharide stachyose and the challenging case of norcamphor, for the latter achieving measurement of all JHH couplings, while only a few were accessible with PSYCHEDELIC. The new experiments are applicable to any organic compound and will prove valuable for configurational and conformational analyses.

| Published April 26, 2024 | DOI: 10.1021/acs.analchem.4c00315

The use of variable domain of the heavy-chain of the heavy-chain-only antibodies (VHHs) as disease-modifying biomolecules in neurodegenerative disorders holds promises, including targeting of aggregation-sensitive proteins. Exploitation of their clinical values depends however on the capacity to deliver VHHs with optimal physico-chemical properties for their specific context of use. We described previously a VHH with high therapeutic potential in a family of neurodegenerative diseases called tauopathies. The activity of this promising parent VHH named Z70 relies on its binding within the central region of the tau protein. Accordingly, we carried out random mutagenesis followed by yeast two-hybrid screening to obtain optimized variants. The VHHs selected from this initial screen targeted the same epitope as VHH Z70 as shown using NMR spectroscopy and had indeed improved binding affinities according to dissociation constant values obtained by surface plasmon resonance spectroscopy. The improved affinities can be partially rationalized based on three-dimensional structures and NMR data of three complexes consisting of an optimized VHH and a peptide containing the tau epitope. Interestingly, the ability of the VHH variants to inhibit tau aggregation and seeding could not be predicted from their affinity alone. We indeed showed that the in vitro and in cellulo VHH stabilities are other limiting key factors to their efficacy. Our results demonstrate that only a complete pipeline of experiments, here described, permits a rational selection of optimized VHH variants, resulting in the selection of VHH variants with higher affinities and/or acting against tau seeding in cell models.
Open Access | Published March 12, 2024 | DOI: https://doi.org/10.1016/j.jbc.2024.107163



The human Mediator complex subunit MED25 binds transactivation domains (TADs) present in various cellular and viral proteins using two binding interfaces, named H1 and H2, which are found on opposite sides of its ACID domain. Here, we use and compare deep learning methods to characterize human MED25–TAD interfaces and assess the predicted models to published experimental data. For the H1 interface, AlphaFold produces predictions with high-reliability scores that agree well with experimental data, while the H2 interface predictions appear inconsistent, preventing reliable binding modes. Despite these limitations, we experimentally assess the validity of MED25 interface predictions with the viral transcriptional activators Lana-1 and IE62. AlphaFold predictions also suggest the existence of a unique hydrophobic pocket for the ArabidopsisMED25 ACID domain.
| Published March 4, 2024 |DOI: https://doi.org/10.1002/1873-3468.14837


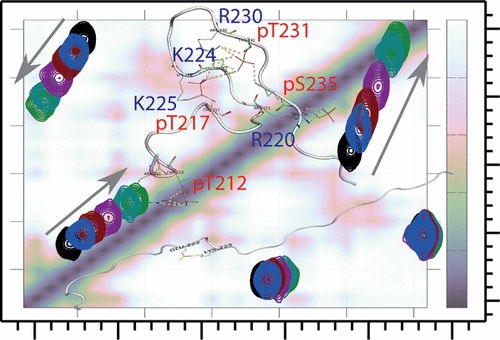
An increase in phosphorylation of the Tau protein is associated with Alzheimer’s disease (AD) progression through unclear molecular mechanisms. In general, phosphorylation modifies the interaction of intrinsically disordered proteins, such as Tau, with other proteins; however, elucidating the structural basis of this regulation mechanism remains challenging. The bridging integrator-1 gene is an AD genetic determinant whose gene product, BIN1, directly interacts with Tau. The proline-rich motif recognized within a Tau(210–240) peptide by the SH3 domain of BIN1 (BIN1 SH3) is defined as 216PTPP219, and this interaction is modulated by phosphorylation. Phosphorylation of T217 within the Tau(210–240) peptide led to a 6-fold reduction in the affinity, while single phosphorylation at either T212, T231, or S235 had no effect on the interaction. Nonetheless, combined phosphorylation of T231 and S235 led to a 3-fold reduction in the affinity, although these phosphorylations are not within the BIN1 SH3-bound region of the Tau peptide. Using nuclear magnetic resonance (NMR) spectroscopy, these phosphorylations were shown to affect the local secondary structure and dynamics of the Tau(210–240) peptide. Models of the (un)phosphorylated peptides were obtained from molecular dynamics (MD) simulation validated by experimental data and showed compaction of the phosphorylated peptide due to increased salt bridge formation. This dynamic folding might indirectly impact the BIN1 SH3 binding by a decreased accessibility of the binding site. Regulation of the binding might thus not only be due to local electrostatic or steric effects from phosphorylation but also to the modification of the conformational properties of Tau.
⏐ Published: May 11, 2023 ⏐ DOI: https://dx.doi.org/10.1021/acs.biochem.2c00717


Deciphering the Structure and Formation of Amyloids in Neurodegenerative Diseases With Chemical Biology Tools
Protein aggregation into highly ordered, regularly repeated cross-β sheet structures called amyloid fibrils is closely associated to human disorders such as neurodegenerative diseases including Alzheimer’s and Parkinson’s diseases, or systemic diseases like type II diabetes. Yet, in some cases, such as the HET-s prion, amyloids have biological functions. High-resolution structures of amyloids fibrils from cryo-electron microscopy have very recently highlighted their ultrastructural organization and polymorphisms. However, the molecular mechanisms and the role of co-factors (posttranslational modifications, non-proteinaceous components and other proteins) acting on the fibril formation are still poorly understood. Whether amyloid fibrils play a toxic or protective role in the pathogenesis of neurodegenerative diseases remains to be elucidated. Furthermore, such aberrant protein-protein interactions challenge the search of small-molecule drugs or immunotherapy approaches targeting amyloid formation. In this review, we describe how chemical biology tools contribute to new insights on the mode of action of amyloidogenic proteins and peptides, defining their structural signature and aggregation pathways by capturing their molecular details and conformational heterogeneity. Challenging the imagination of scientists, this constantly expanding field provides crucial tools to unravel mechanistic detail of amyloid formation such as semisynthetic proteins and small-molecule sensors of conformational changes and/or aggregation. Protein engineering methods and bioorthogonal chemistry for the introduction of protein chemical modifications are additional fruitful strategies to tackle the challenge of understanding amyloid formation.
Open Access ⏐ Published: May 12, 2022 ⏐ DOI: https://doi.org/10.3389/fchem.2022.886382


Inhibition of Tau seeding by targeting Tau nucleation core within neurons with a single domain antibody fragment
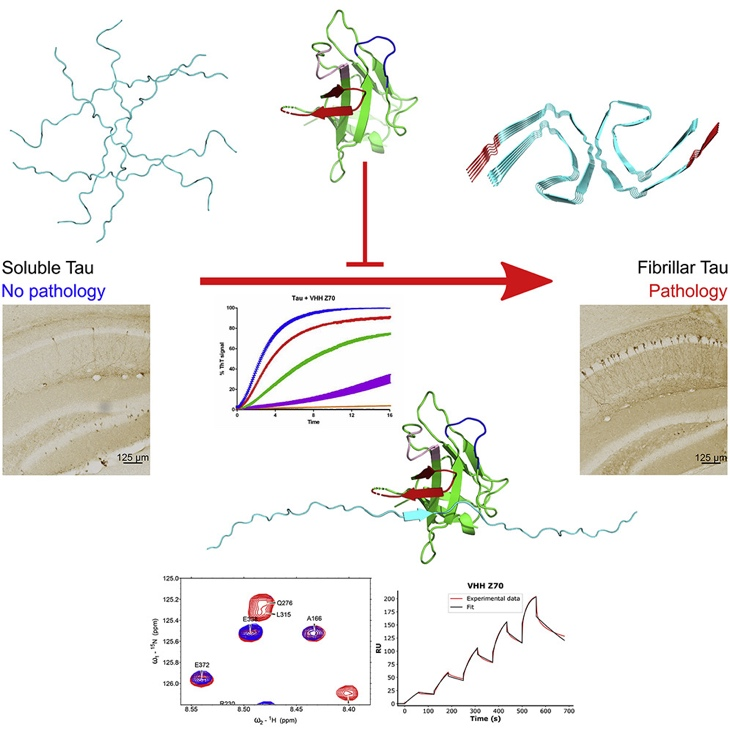
Tau proteins aggregate into filaments in brain cells in Alzheimer’s disease and related disorders referred to as tauopathies. Here, we used fragments of camelid heavy-chain-only antibodies (VHHs or single domain antibody fragments) targeting Tau as immuno-modulators of its pathologic seeding. A VHH issued from the screen against Tau of a synthetic phage-display library of humanized VHHs was selected for its capacity to bind Tau microtubule-binding domain, composing the core of Tau fibrils. This parent VHH was optimized to improve its biochemical properties and to act in the intra-cellular compartment, resulting in VHH Z70. VHH Z70 precisely binds the PHF6 sequence, known for its nucleation capacity, as shown by the crystal structure of the complex. VHH Z70 was more efficient than the parent VHH to inhibit in vitro Tau aggregation in heparin-induced assays. Expression of VHH Z70 in a cellular model of Tau seeding also decreased the aggregation-reporting fluorescence signal. Finally, intra-cellular expression of VHH Z70 in the brain of an established tauopathy mouse seeding model demonstrated its capacity to mitigate accumulation of pathological Tau. VHH Z70, by targeting Tau inside brain neurons, where most of the pathological Tau resides, provides an immunological tool to target the intra-cellular compartment in tauopathies.
Open Access ⏐ Published: January 06, 2022 ⏐ DOI: https://doi.org/10.1016/j.ymthe.2022.01.009


Institut Pasteur de Lille research highlight: The S100B protein protects the brain from toxic protein Tau aggregates
https://pasteur-lille.fr/a-la-une/actualites-scientifiques/#tau



Dynamic interactions and Ca2+-binding modulate the holdase-type chaperone activity of S100B preventing tau aggregation and seeding
The microtubule-associated protein tau is implicated in the formation of oligomers and fibrillar aggregates that evade proteostasis control and spread from cell-to-cell. Tau pathology is accompanied by sustained neuroinflammation and, while the release of alarmin mediators aggravates disease at late stages, early inflammatory responses encompass protective functions. This is the case of the Ca2+-binding S100B protein, an astrocytic alarmin which is augmented in AD and which has been recently implicated as a proteostasis regulator, acting over amyloid β aggregation. Here we report the activity of S100B as a suppressor of tau aggregation and seeding, operating at sub-stoichiometric conditions. We show that S100B interacts with tau in living cells even in microtubule-destabilizing conditions. Structural analysis revealed that tau undergoes dynamic interactions with S100B, in a Ca2+-dependent manner, notably with the aggregation prone repeat segments at the microtubule binding regions. This interaction involves contacts of tau with a cleft formed at the interface of the S100B dimer. Kinetic and mechanistic analysis revealed that S100B inhibits the aggregation of both full-length tau and of the microtubule binding domain, and that this proceeds through effects over primary and secondary nucleation, as confirmed by seeding assays and direct observation of S100B binding to tau oligomers and fibrils. In agreement with a role as an extracellular chaperone and its accumulation near tau positive inclusions, we show that S100B blocks proteopathic tau seeding. Together, our findings establish tau as a client of the S100B chaperone, providing evidence for neuro-protective functions of this inflammatory mediator across different tauopathies.